

The highly pathogenic H5N1 avian influenza epidemic that occurred between 2021 and 2022in North-Eastern Italy was one of the most serious ever recorded, involving 317 poultry farms and resulting in the culling of over 14 million birds. Most of the affected farms were located in an area considered highly densely populated, where approximately 70% of the Italian poultry is reared. The spread of the epidemic was extremely rapid, initially affecting the province of Verona and then extending to the surrounding provinces and regions, with peaks of over 50 new outbreaks per week.
Two main hypotheses were proposed to account for the rapid spread of the epidemic in the area: 1) direct contacts may have occurred among infected farms and other poultry companies, or 2) common sources of infection may have been present, leading to multiple new outbreaks over a short period of time.

The National Reference Centre for Avian Influenza & Newcastle Disease of the IZSVe addressed the challenge of explaining the transmission dynamics of the 2011-2012 avian influenza epidemic by investigating the impact of factors potentially associated with the spread of infection. The analytical approach used by the research team belongs to network analysis, a set of integrated techniques applicable in various fields, including economic, social sciences, psychology, and biology.
The Laboratory of Epidemiology and Risk Analysis in Public Health (SCS4) of the Istituto Zooprofilattico Sperimentale delle Venezie (IZSVe) addressed the challenge of explaining the transmission dynamics of the epidemic by investigating the impact of factors potentially associated with the spread of infection. The analysis was based on data collected during on-site epidemiological investigations of the affected farms and genetic information yielded by biomolecular analyses on the viruses isolated from each outbreak site. The study was published in the journal Pathogens.
Network analysis and the Avian Influenza Epidemic of 2021-2022
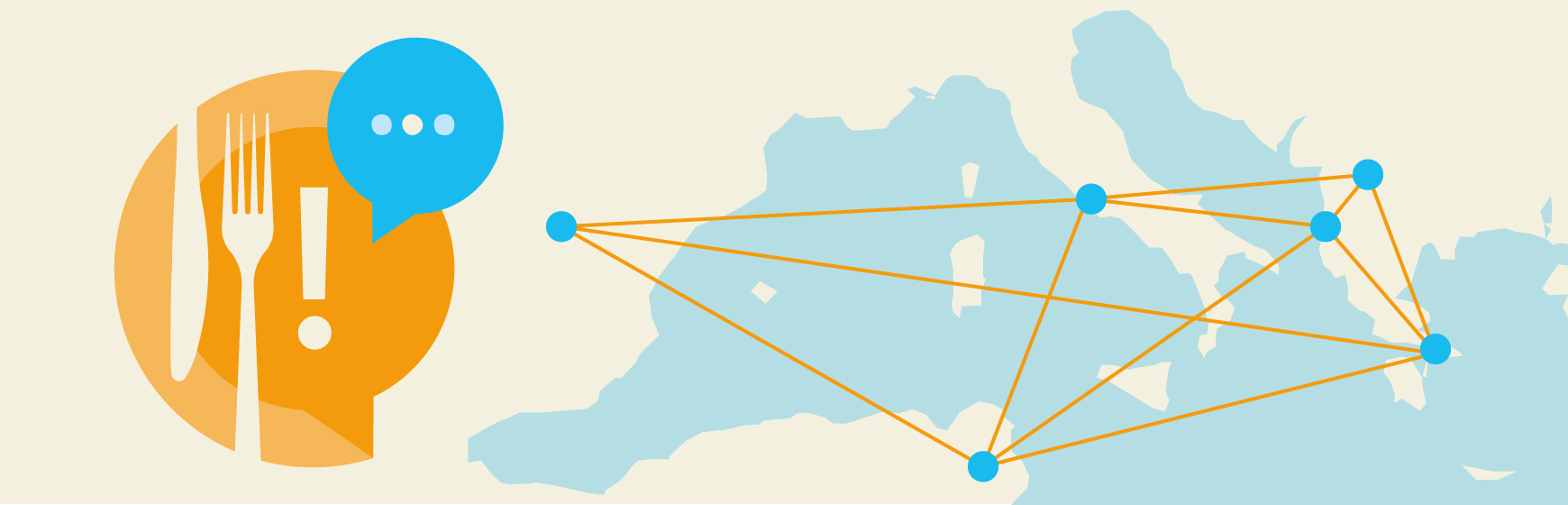
The analytical approach used by the research team belongs to network analysis, a set of integrated techniques applicable in various fields, including economic, social sciences, psychology, and biology. This powerful analytical tool is designed to explore the characteristics and relationships among objects within a network: the nodes represent entities, such as objects or actors, while the links represent relationships among those entities.
In the context of the 2021-2022 epidemic, phylogenetic analyses performed by the National Reference Centre for Avian Influenza & Newcastle Disease of IZSVe revealed the existence of various genetic clusters of viruses, supporting the hypothesis of viral transmission among the domestic farms. Subsequent virological analyses then focused on assessing the genetic similarity of the sequenced viruses belonging to the same cluster.
Specifically, a phylogenetic network was built using the complete genomes of 214 viral strains. In this network, each node represented a strain identified in a single outbreak, while the links connected the nodes showing the highest genetic similarity. The phylogenetic network served as the baseline data source used for constructing an Exponential Random Graph Model (ERGM). ERGM is a statistical model in network analysis that explain why a link exists between two nodes based on a series of epidemiological variables. When applied to the phylogenetic network, the ERGM identified the most significant epidemiological characteristics of the affected farms and evaluated their impact on the probability of observing a link within the genomic network, that is, the genetic similarity of the isolated viral strains, and thus the potential spread of the infection among farms.
Network variables underlying viral transmission

The analyses revealed that certain variables played a significant role in defining the network structure and, consequently, in potential disease transmission. The most important drivers were related to belonging to the same poultry company, the time of exposure to active outbreaks, and the geographical distance between farms, raising important implications for potential control, management, and prevention of future avian influenza outbreaks in northeastern Italy.
The analyses revealed that certain variables played a significant role in defining the network structure and, consequently, in potential disease transmission. The most important drivers were related to belonging to the same poultry company, the time of exposure to active outbreaks, and the geographical distance between farms, raising important implications for potential control, management, and prevention of future avian influenza outbreaks in northeastern Italy.
The innovative application of network analysis techniques – typically used in the context of social sciences – to study the avian flu epidemic proved to be an effective method for integrating virological and epidemiological data. Unlike most scientific papers published in the literature, where epidemiological and genomic aspects are usually presented separately, in this study the epidemiological data were directly used to ‘explain’ the structure of the virological data.
Further developments in this approach are in progress. For example, information related to the evolution of an epidemic over time can be integrated through ‘temporalized’ networks, allowing for a more detailed understanding of the spatiotemporal transmission dynamics of a given infection. Similarly, diseases affecting various populations (such as vector-borne diseases) could be studied through multi-layer networks, which add a higher level of complexity related to the presence of several entities (called ‘layers’) interacting within the same system.
The potential application of network analysis in the future could assist epidemiologists in better understanding the complexity of diseases transmission and circulation and provide an effective tool for studying, monitoring and preventing the emergence of new epidemics.
Read the scientific article in Pathogens »

{kind=link}
{kind=link}
{kind=link}
{kind=link}